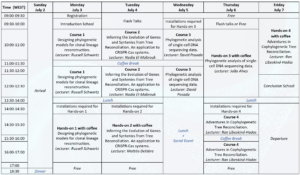
Agenda
Lecturers and Hands-on


David Posada, University of Vigo in Galicia, NW Spain
Title: Phylogenetic analysis of single-cell DNA sequencing data.
Abstract: Concepts and techniques from organismal evolutionary biology can provide a detailed, quantitative picture of the complex dynamics of somatic cell populations over time and space. In the theoretical lecture, we reviewed basic concepts of cancer evolution, the complexity of single-cell DNA sequencing data (scDNA-seq), different phylogenetic techniques for analyzing these types of data, together with empirical examples of their application. During the hands-on practical session, participants acquired basic skills to perform scDNA-seq phylogenetic analysis with CellPhy, including variant calling, setting up of genotype models, tree search, bootstrap support estimation, and mutation mapping. Participants learnt to prepare their data, explore its characteristics, and present their results using tree plotting packages in R.
Biosketch: David Posada (Vigo, 1971) is Professor of Genetics at the University of Vigo (Spain). After graduating in Biology from the University of Santiago de Compostela (Spain), he received his doctorate in USA, where he also completed his postdoctoral training at a pharmacogenomics company in Boston (Variagenics Inc) and at the MIT. His work is framed within the field of molecular evolution and computational biology, areas in which he has received more than 70,000 citations. His current interests focus on tracing the evolutionary history of tumors, for which he leverages single-cell genomics, evolutionary biology and computational phylogenomics.
The Hands-on Session was given by
João Alves, University of Vigo
Biosketch:
I received my PhD at the Instituto de Ciências Abel Salazar (University of Porto, Portugal) in 2014, where I studied the impact of a particular subtype of structural variant – chromosomal inversions – on the evolution of the human genome. Currently, my research is broadly focused on incorporating population genomics strategies to explore cancer evolution and progression, as well as to understand the underlying causes of somatic variation within tumors (i.e., intratumor heterogeneity). In addition, I have been increasingly interested in evaluating the prognostic potential of genomic heterogeneity and tumor mutational burden in predicting cancer-specific survival.


Nadia El-Mabrouk, Université de Montréal
Title: Inferring the Evolution of Genes and Syntenies from Tree Reconciliation. An application to CRISPR-Cas systems.
Abstract: In this course, I introduced algorithmic results for inferring the evolution of a gene family or a set of co-localized gene families in a genome. After a brief review of species tree/gene tree reconstruction methods and challenges, I introduced the reconciliation approach which, given a rooted (resolved or partially resolved) gene tree and a given rooted and binary species tree, predicts an embedding of the gene tree into the species tree inducing a most parsimonious (or more likely) duplication/loss/gain/transfer scenario for the gene family. In a second part, I addressed the problem of inferring the evolution of “syntenies”, i.e., groups of co-localized genes evolving together from an ancestral genomic segment, considering or ignoring the order of genes. I showed how the reconciliation method can be generalized to a synteny tree. The application part of the presentation was addressed the case of the CRISPR–Cas module, an adaptive system used by microbes to defend against invading viruses and plasmids, leading to the most reliable and accurate “molecular scissors” with important biotechnology and biomedical applications. The study and analysis of CRISPR-Cas systems have revealed a remarkable wide diversity of Cas protein sequences, but also composition and architecture. We will consider the evolution of Class I CRISPR-Cas systems composed of multi-subunit effector proteins. We took this example as a case study and, in the hands-out sessions, addressed the practical questions of isolating Cas genes, dealing with duplicates, constructing gene trees, inferring synteny trees and running the reconciliation tool.
Biosketch: Nadia El-Mabrouk is full professor at the Computer Science Department and member of the «Centre de Recherche Mathématiques» at the University of Montreal. She has a longstanding experience in developing algorithms for comparative genomics and especially genome rearrangements, gene tree reconstruction and Gene tree/Species tree reconciliation. She is involved, each year, in the program committee of some of the most popular conferences in computational biology such as ISMB/ECCB, RECOMB, WABI and APBC. She acted as the ISMB Proceeding Co-chair in 2018 and 2019 and has since been co-chairing the Evolutionary and Comparative Genomics COSI at ISMB. She organized two RECOMB Comparative Genomics Workshops in Montreal. Her research appears in a variety of computer science, bioinformatics and life science journals, among them IEEE/ACM, Molecular Biology and Evolution, Bioinformatics, Nature Scientific Reports and BMC-Genomics.
In this case, the Hands-on Session will be given by
Mattéo Delabre, Université de Montréal
Biosketch: TBA

Ran Libeskind-Hadas, Claremont McKenna College
Title: Adventures in Cophylogenetic Tree Reconciliation.
Abstract: The talks and hands-on sessions explored current methods for reconciling pairs of phylogenetic trees to provide insights into the co-evolutionary histories of pairs of taxa. We discussed different approaches to tree reconciliation and their relative merits. In particular, we described the Jane and eMPRess software tools in some detail, their underlying algorithms, their strengths, and their limitations. In the hands-on sessions, participants used these tools on a number of datasets – including their own datasets if they wish.
Biosketch: Ran “RON” Libeskind-Hadas received his undergraduate degree in applied mathematics from Harvard University and the MS and PhD from the University of Illinois at Urbana-Champaign. Subsequently, he was on the faculty at Harvey Mudd College where he served as department chair in the Department of Computer Science and Associate Dean of the College. In 2002, he moved to Claremont McKenna College as Founding Chair of the Kravis Department of Integrated Sciences. His research interests are in algorithmic computational biology and, in particular, in coevolutionary analyses via reconciliation of phylogenetic trees (e.g, host and symbiont, host and parasite, or species and gene trees). His research group developed the widely-used Jane reconciliation tool and, more recently, the eMPRess tool. He is the co-author of the textbook “Computing for Biologists” and a contributor to the book “Bioinformatics for Biologists” (both from Cambridge University Press).

Russell Schwartz, Computational Biology Department, Carnegie Mellon University
Title Talk: Designing phylogenetic models for clonal lineage reconstruction.
Abstract: These sessions explored how we can design our own phylogenetic models and adapt them to different scenarios, data types, and mutation models, with application to problems in clonal phylogenetics and deconvolution in cancers. The lecture material provided background information on clonal deconvolution and phylogenetics and used these to illustrate principles in computationally modeling an optimization problem. We then explored techniques for implementing these problem statements through the technique of integer linear programming. Hands-on sessions worked with Jupyter Notebooks and R to allow us to implement some simple phylogenetic models and explore how we can generalize and adapt them to new versions of the inference problem.
Biosketch: Russell Schwartz has been a faculty member of Carnegie Mellon University since 2002, where he is currently Professor and Head of the Computational Biology Department and Professor of Biological Sciences with additional appointments in the Computer Science Department and Machine Learning Department. His laboratory has worked broadly on algorithms, machine learning, and simulation methods for computational genetics, genomics, and biophysics, with a current focus largely on computational cancer biology and somatic variation. He is also active in bioinformatics education, largely through work with the International Society for Computational Biology (ISCB), where he currently serves as a Vice President and as co-chair of its Education Community of Special Interest (COSI).
Registration
The registration fee was 250€ for PhD students and members of the OLISSIPO project, 350€ for postdocs or other researchers (meals indicated at the schedule of the school are included, accommodation and flights are not).
Flash Talks
We had slots for flash talks (3-10 min) to ESRs present the work they have been developing in their research.
Organizers
Susana Vinga (INESC-ID), Marie-France Sagot (Inria), Niko Beerenwinkel (ETH Zürich), Wolfgang Huber (EMBL), Blerina Sinaimeri (Inria and LUISS University), Alexandre Francisco (INESC-ID/IST), João Carriço (BioMérieux) and Sara Tanqueiro (INESC-ID)
Photos














Funding
The H2020 Twinning Project Olissipo has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 951970.